The In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746 has been in effect since May 26, 2022. The IVDR replaced the In Vitro Diagnostic Medical Devices Directive (IVDD). The IVDR establishes a new classification system for diagnostics and requires a conformity assessment from a Notified Body. It also requires manufacturers to report serious incidents and trends in non-serious incidents. The IVDR applies to all 27 European Union member states, Iceland, Liechtenstein, Norway, and Switzerland.
The IVDR does not apply to Great Britain (GB), which is part of the United Kingdom (UK). The IVDR did not take effect during the Brexit transition period, which ended on December 31, 2020. GB continues to follow the rules set out in the UK Medical Devices Regulations 2002 (UK MDR 2002). Northern Ireland has a special status and continues to follow EU rules, including the IVDR. The UK has a transitional period for IVDs that comply with EU rules to be placed on the market in GB until June 30, 2030. After that date, such IVDs will need to bear a UKCA mark. The UK Medicines and Healthcare products Regulatory Agency (MHRA) is responsible for regulating medical devices in the UK.
The IVDR also allows health institutions to manufacture, modify, and use devices in-house to meet the needs of their patients. By the end of the transition period, all IVD devices – new and existing – will have to be reclassified and will require Notified Bodies review. Therefore, the vast majority of the previously self-declared industry would require a notified body to certify their products and must undergo extensive changes in the systems and processes, as new classification rules are applied to all CE-marked products.
IVD Definition
IVD stands for in vitro diagnostics, which are tests that analyze samples from the human body to detect diseases, conditions, or infections. The term “in vitro” means “in glass,” and these tests are typically performed in test tubes or other similar equipment.
IVDs can be used to:
- Diagnose patients.
- Monitor a person’s health.
- Help cure, treat, or prevent diseases.
- Identify patients who are likely to benefit from specific treatments.
IVDs can be performed in a variety of settings, including laboratories, health care facilities, or at home. The tests can be performed using a range of instruments, from small, handheld tests to complex laboratory instruments.
The FDA classifies IVDs into Class I, II, or III, based on the level of regulatory control needed to ensure their safety and effectiveness. Class I IVDs have the least regulatory control, while Class III IVDs have the most stringent requirements.
IVDR Suitability
IVDR Articles 2(2), 2(3) define in vitro diagnostic medical device as “any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.”
IVDR also includes Specimen receptacles in its definition: “specimen receptacle means a device, whether of a vacuum-type or not, specifically intended by its manufacturer for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination;“
IVDR Classification
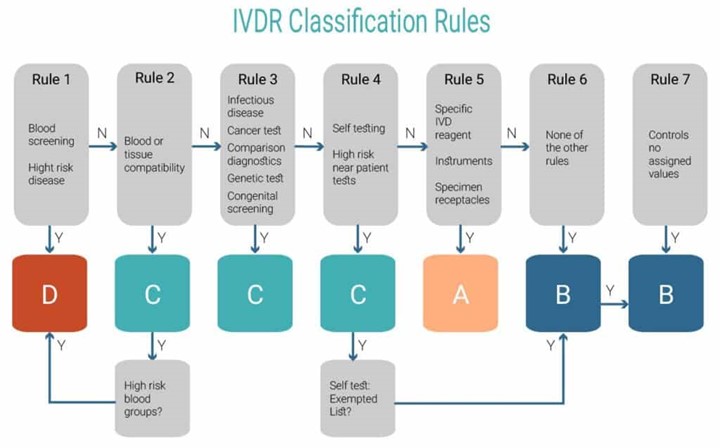
Annex VIII defines the classification rules needed for reclassifying a device. This process is mandatory and is the initial “gateway” for downstream activities concerning all aspects of R&D, RA and QA. IVD classification under IVDR is similar to medical device classification under Medical Device Reporting (MDR) per 21 CFR Part 803 in the sense that it addresses device classes according to the risk they pose. The IVDR classification rules include 5 classes of devices: A, A sterile, B, C and D. Class A (non-sterile) manufacturers can remain self-declared, however, all other IVDs A sterile – D require Notified Body approval.
IVD posed risk is measured on two scales: personal risk and public health risk. The risk to public health is considered as the more critical risk. Meaning, that IVDs with even high personal risk may be classified only as high as class C as long as they don’t pose a high public health risk, whereas a mirroring IVD will be the highest class – D. The new, rule-based IVDR classification system for devices makes the IVDR more practical, by allowing it to remain relevant to a changing industry.
- Class A sterile – low personal risk and low public health risk. Class A sterile IVDs only require Notified Body approval for the sterile part (the non-sterile is self-declared class A).
- Class B – low/moderate personal risk and low public health risk.
- Class C – high personal risk and low/moderate public health risk.
- Class D – high personal risk and high public health risk.
The IVDR Classification Rules are described in the figure below. Classification according to Annex VIII runs from the most severe to the least severe of devices: high public health risk is class D, high personal risk is class C, and so on. An IVD not falling under any specifically mentioned category is class B.