
ISO 13485 is the internationally recognized quality management benchmark for medical device manufacturers. Getting ISO 13485 certified conveys trust of by regulators, stakeholders, and customers while clearing the path market. ISO 13485:2016 offers benefits:
- Brings quality and continuous improvement into the medical device organization;
- Improved patient/customer satisfaction by consistently providing safe medical devices meeting customer requirements;
- Enhanced reputation and credibility;
- Greater efficiency;
- Reduced costs due to reduced waste, rework and other inefficiencies;
- Improved risk management; and
- A stronger foundation for growth to expand into new markets.
ISO 13485 is an internationally agreed upon set of standard quality management system (QMS) requirements for companies involved in the design, production, installation, servicing, and manufacturing of medical devices. ISO 13485 was first published in 1996 and has since been revised in 2003 and 2016. The current version, ISO 13485:2016, came into effect in March 2016. The goal of the requirements is to ensure that medical devices and services consistently meet customer expectations and relevant regulatory requirements. The ‘ISO’ in ISO 13485 stands for the International Organization for Standardization, which publishes the international standards governing most modern industries. The ‘13485’ is the designated numerical code given to ISO’s medical device quality management standard. ISO 13485 certification is provided to any medical device organization that meets the requirements of ISO 13485.
ISO is an international non-governmental organization of industry leaders who share their knowledge and expertise to provide solutions for global challenges. ISO 13485 covers ISO 9001 with a few additional requirements. Consumers and the life science supply chain have come to trust ISO, and they’ll often refuse to purchase medical device products from companies that lack ISO 13485 certification. To obtain CE marking—which indicates conformity with safety standards for products sold in the European Economic Area—medical device manufacturers must get ISO 13485 certified with a notified body and have a Quality Management System (QMS) in place. ISO 13485 has also taken on additional significance in the United States in recent years, as the FDA plans to harmonize its own 21 CFR 820 medical device quality requirements with those of ISO 13485. The ISO 13485 vs 21 CFR 820 comparison which American medical device manufacturers have had to work through in the past looks set to be broken down when the FDA’s new QMSR goes live in 2026 – making ISO 13485 best practice a key part of American national regulatory expectations.
ISO 13485 certification cost varies depending on the size and complexity of the organization and product. Fees are paid directly to the notified body conducting the ISO 13485 assessment. Around $20,000 is a minimum. This includes the typical annual certification fee of $3000-5000, typical audit costs of around $3000 per day, plus any other billable planning and reporting time and the associated work and time expenses of the company internal preparation work.
ISO 13485 includes requirements for design and development, risk management, production, and post-production processes for medical device companies. Below are five key requirements from ISO 13485:
1. Quality Management System (QMS)
To be certified to ISO 13485, a company must implement and maintain a quality management system meeting the requirements set out in the standard. According to ISO, organizations need to:
- Determine the processes the quality management system requires and what’s needed to apply these processes throughout the organization, taking into account the various roles involved;
- Apply a risk-based approach to the control of the appropriate processes needed for the quality management system; and
- Determine the sequence and interaction of these processes.
2. Management Responsibility
Management should provide evidence of its commitment to the development and maintenance of the quality management system and its effectiveness. To do that:
- Communicate the importance of meeting regulatory requirements;
- Establish high-value quality policy;
- Ensure quality objectives are established;
- Conduct management reviews; and
- Ensure availability of quality management system resources.
3. Resource Management
Resources should include:
- Human resources;
- Infrastructure;
- Work environment; and
- Contamination control.
Product Realization
- Establish the quality requirements for the product(s);
- Define the required processes and what supporting documentation will be needed for those processes;
- Outline the company infrastructure that will need to be created and what the work environment should be like;
- Define employee qualification and training requirements;
- Establish processes for verification, validation, measurement, monitoring, handling, inspection, storage, distribution, and traceability;
- Organize the information and include measurement, analysis, and improvement.
According to ISO, “the organization shall plan and implement the monitor, measurement, analysis, and improvement processes” related to the quality management system and products. So, organizations need to:
- Demonstrate conformity of product;
- Ensure conformity of the quality management system; and
Maintain the effectiveness of the quality management system.
ISO 13485 QMS Development Diagram
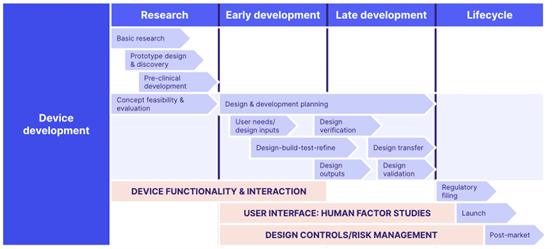
ISO 13485 contains 8 clauses as part of its requirements:
1. Scope
The scope sets out the intended outcomes of the modern medical device quality management system, including the significance of the process approach and continuous improvement.
Normative References-Provides details of the reference standards or publications relevant to the particular standard, including ISO 9001:2015.
3. Terms & Definitions
Details terms and definitions applicable to the standard, including definitions of Active Implantable Medical Device, Active Medical Device, Advisory Notice, Customer Complaint, Implantable Medical Device, Labeling, Medical Device, and Sterile Medical Device.
4. General requirements
Lays out the broad requirements for a properly documented ISO13485 QMS, including:
- Quality manual with clear QMS scope;
- Documentation control procedures; and
- Required forms, records and SOPs.
5. Management responsibility
Pertains to the role of ‘top management’ or the group of people who direct and control the organization at the highest level. Customer and patient satisfaction and safety should be overseen and maintained by top management with:
- Clear responsibilities;
- Frequent management reviews; and
- A clear quality policy with objectives.
6. Resource management
Requirements for how resources are managed and applied to meet the quality objectives, including personnel, equipment, and training.
7. Product realization
Maps the requirements for the end-to-end medical device product realization process, including:
- Production and manufacture;
- Capturing and actioning feedback;
- Planning;
- Design;
- Purchasing; and
- Traceability.
8. Measurement, analysis, and improvement
Breaks down how to monitor and analyze the processes with a view to continuous refinement and improvement. Core considerations include:
- Auditing;
- CAPAs;
- Non-conformance control; and
- Measuring and maximizing customer satisfaction and patient/product safety.
In preparation for an ISO 1345 audit, best practices include:
- Device master record explicitly defining QMS requirements. The ISO 13485 medical device file is a key document needed to demonstrate compliance to the standard. The MDF should document the device’s design, development, and testing activity to prove that it works as intended. It should also include the risk management activities, as well as any post-market surveillance data once the device is in the market.
- Feedback and review system for non-conformance detection;
- Product quality control (monitoring and measuring) throughout production process;
- Set quality requirements must be met before product release and delivery;
- Advisory notices, rework activity, release of non-conforming product (which still meets regulatory requirements) must be documented;
- Personnel require access to procedures, requirements; and reference materials at the point of work;
- Unique and specific records for every approved and verified device batch;
- Installation and verification device requirements;
- Maintained records of device installation, verification and servicing activities and procedures;
- QMS containing product specification documents and quality policy, with a framework for reviews and updates controlled by the management team;
- Management must verify QMS goals and compliance;
- Documented procedures for shelf life, quality data collection/analysis/ retention, maintenance activity, risk/environment management, adverse event flagging, product conformity, identification, returns, maintenance, labeling and packaging.
- Ensure CAPA standards are met-Refer to the FDA’s inspection guidelines and to ISO 13485 8.5.3 (prevention) and ISO 13485 8.5.2 (correction);
- Implement complaint procedures-Establish complaint procedures following the guidelines laid out in FDA CFR 820.198 and ISO 13485 8.2.2. A lack of standard procedures for handling complaints or failure to provide evidence that they followed procedures is the second most common reason organizations received a 483 observation;
- Include purchasing controls-Create a written procedure for supply chain management to reduce the risk of noncompliance or supplier risks that could compromise your device quality.
- Develop MDR procedures-MDR (Medical Device Reporting) should include events and annual reports as detailed under FDA CFR 803.17 and ISO 13485:2016. Written procedures and systems are critical for compliance with record-keeping guidelines for MDR;
- Create a process to prepare for the audit-Review the following areas every three months: Design, Trainings, Purchasing, and Quality assurance;
- Focus on upstream quality-Manufacturers use the term “Upstream Quality (UQA)” to refer to a concept that relates to quality from the start. Focusing on UQA means putting effort into planning in the early stages to reduce quality issues later down the line;
- An eQMS -Electronic quality management systems designed for life sciences companies are built using the ISO 13485 framework for quality control, operational efficiency, regulatory compliance, and the safe manufacture of medical devices. The eQMS should provide essential functions such as document control, training, and the ability to expand to other areas — like CAPA. The eQMS is essential for risk management, testing, and other procedures to streamline product submission.